bulletin | feature story
Decoding the structural genome of silicate glasses
By Qi Zhou, Mathieu Bauchy, and Ying Shi
Silicate glasses can exhibit a wide range of properties.
To understand, tune, and enhance the properties of silicate glasses, one needs to decode the “glass genome,” that is, to uncover how basic structural features control a glass’s macroscopic properties.1,2 Such decoding requires accurate knowledge of the atomic structure of silicate glasses. However, despite silicate glasses’ ubiquity and technological importance, their atomic structure—especially at the medium-range order—remains only partially understood.3
A 3D rendering of the structure of silica glass.
Credit: Zhou
Here, we present force-enhanced atomic refinement (FEAR) as a powerful modeling technique to unveil the three-dimensional structure of glasses.
Limitations of present experimental techniques
To date, no experimental technique can directly probe the three-dimensional atomic structure of silicate glasses. Conventional experiments solely offer some “fingerprints” of the glass structure—for instance, diffraction experiments and nuclear magnetic resonance can provide the structure factors and coordination numbers. Although this information offers some useful constraints on the nature of the glass structure, it does not directly reveal the three-dimensional structure itself.
Challenges with modeling approaches
As an alternative route to experiments, atomistic simulations offer direct and full access to the atomic structure of glasses. However, atomistic simulations come with their own challenges and limitations.4,5
For example, molecular dynamics (MD) simulations solely rely on knowledge of the interatomic forcefield. Following the melt-quench method, melts are equilibrated at high temperature and subsequently quenched to the glassy state with a high cooling rate. Although this melt-quenching approach roughly mimics the experimental synthesis protocol of glasses, MD simulations are limited to very large cooling rates (typically 10–2 to 102 K/ps) due to their computational cost.5 This limitation is serious because the structure and properties of glasses depend on their thermal history.
An additional example is conventional reverse Monte Carlo (RMC) simulations, which solely rely on knowledge of experimental constraints.6 As a key advantage, RMC simulations can yield glass structures that are compatible with such constraints while bypassing the melt-quenching route, thereby avoiding the issue of the cooling rate. However, an RMC simulation remains an ill-defined approach because, for instance, numerous atomic structures can exhibit the same pair distribution function. As such, glass structures that are generated by RMC typically exhibit an excellent agreement with the experimental data but may nevertheless be fairly unrealistic (e.g., showing extremely high potential energy).7

ADVERTISEMENT
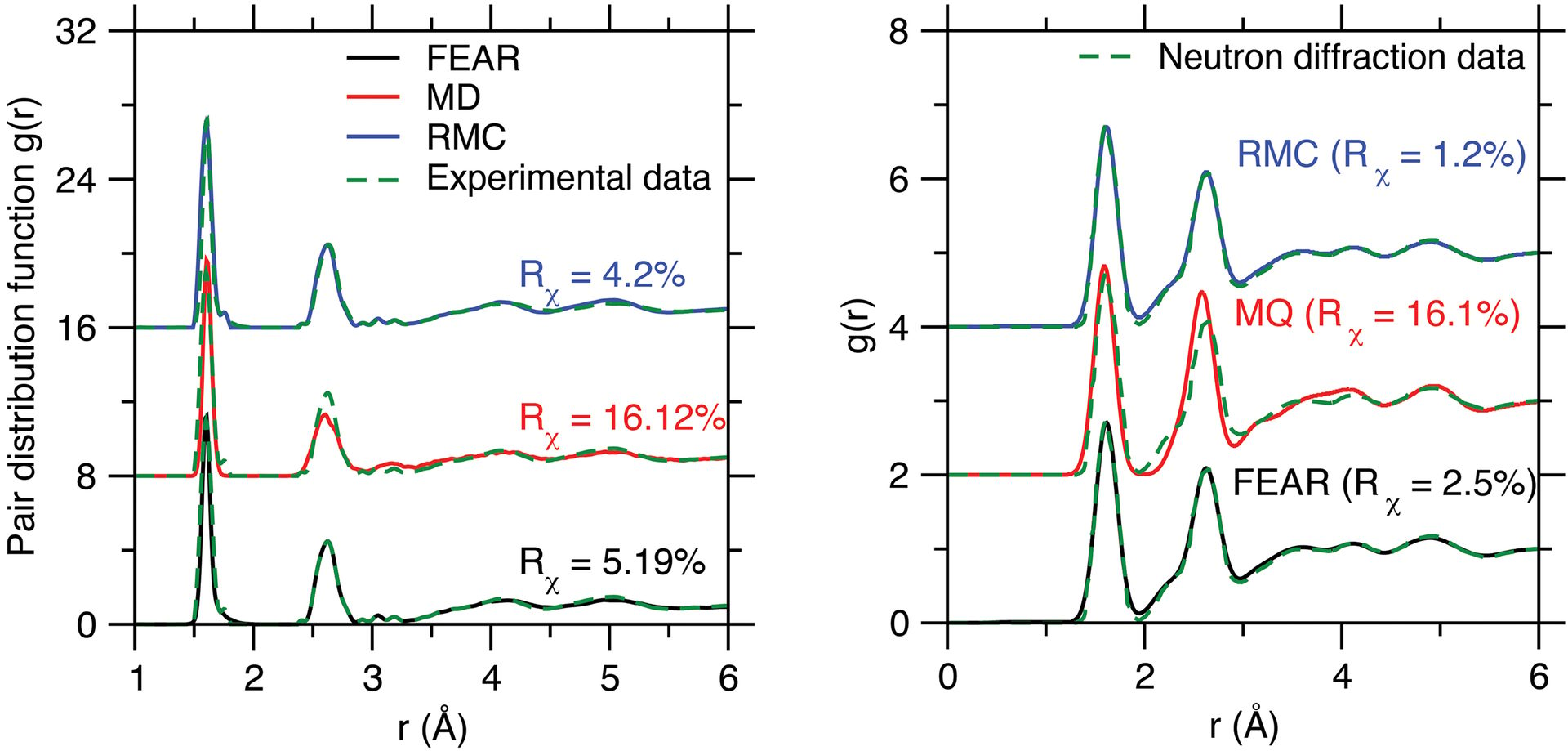
Fig. 2. Neutron pair distribution functions (PDFs) of (left) silica and (right) sodium silicate glasses obtained by force-enhanced atomic refinement (FEAR), molecular dynamics (MD) using the melt-quench (MQ) method (1 K/ps), and reverse Monte Carlo (RMC). All the PDFs are compared with available experimental neutron diffraction data. The silica graph is republished from Ref. 9, while the sodium silicate graph is created from data reported in Ref. 7.
Credit: (left) Zhou et al., J. Non-Cryst. Solids 2021 (CC BY 4.0); (right) Zhou, Shi, and Bauchy
Force-enhanced atomic refinement (FEAR)
To overcome the limitations of MD and RMC, we adopted force-enhanced atomic refinement, or FEAR. This recent method leverages all available information, namely, (i) the interatomic forcefield, which is typically used by MD simulations; and (ii) experimental constraints, which are typically used by RMC simulations.8
In detail, FEAR relies on an iterative combination of sequential energy minimizations and RMC refinements wherein a pair distribution function (PDF) obtained by diffraction is used as the target. Technical details can be found in Refs. 7 and 9.
Quantitative agreement with experimental data
Figure 2 shows the neutron PDFs of silica and sodium silicate glasses. The level of agreement between FEAR and diffraction data is comparable to what is achieved by RMC, which is not surprising because RMC solely aims to minimize the difference between the simulated and experimental PDFs.
On the other hand, the level of accuracy offered by FEAR largely exceeds that of MD. Although MD yields a reasonable description of a glass’s short-range order, the level of agreement between MD and diffraction data is lower at the medium-range order. In contrast, for both glasses, the PDFs of the glass structures generated by FEAR show an excellent agreement with experimental neutron data for both the short- and medium-range length scales.7,9
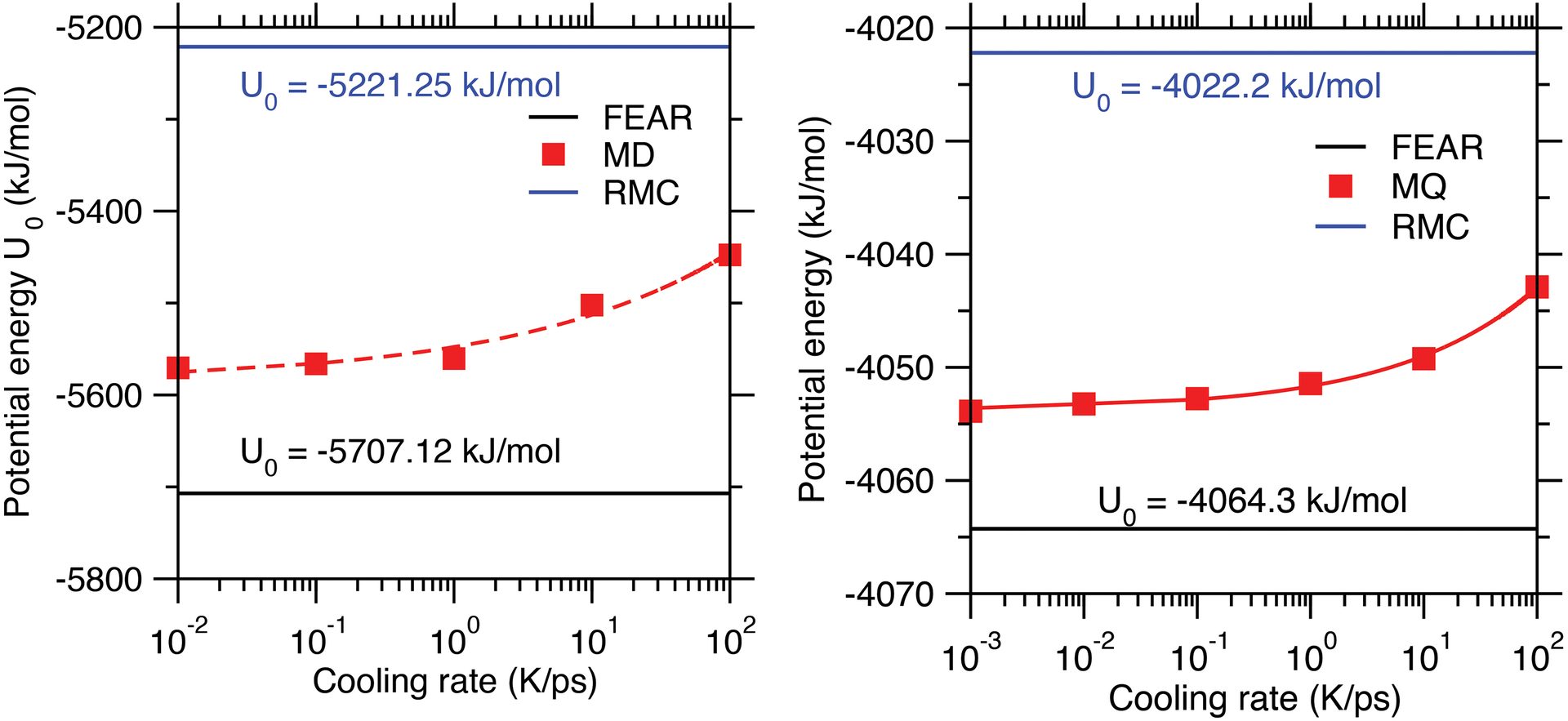
Figure 3. Molar potential energy of melt-quenched (MQ) glasses generated by molecular dynamics (MD) simulations as a function of the cooling rate for (left) silica and (right) sodium silicate glasses. Values obtained for the glasses generated by force-enhanced atomic refinement (FEAR) and reverse Monte Carlo (RMC) are shown as horizontal lines for comparison. The silica graph is republished from Ref. 9, while the sodium silicate graph is created from data reported in Ref. 7.
Credit: (left) Zhou et al., J. Non-Cryst. Solids 2021 (CC BY 4.0); (right) Zhou, Shi, and Bauchy
Unmatched thermodynamic stability
In addition to demonstrating excellent agreement with experimental data, the glass structures generated by FEAR exhibit an unmatched level of thermodynamic stability.
Figure 3 shows the molar potential energy of silica and sodium silicate glass structures generated by FEAR. It can be seen that FEAR yields some potential energies that are significantly lower than those offered by RMC—meaning the FEAR glasses are more thermodynamically stable. The high energy of the RMC structures exemplifies the fact that, although the PDFs calculated from these glass structures offer an excellent match with diffraction data, the configurations yielded by RMC are thermodynamically unstable.
The potential energy of the structures generated by FEAR is also notably lower than those obtained by MD, including for very slow cooling rates. This result demonstrates that, although FEAR and MD rely on the same interatomic forcefield, FEAR allows the simulated glass to reach more stable energy states.
All these results demonstrate that FEAR offers an improved description of the atomic structure of glassy silica as compared to traditional MD simulations based on the melt-quench method or RMC simulations.
Acknowledgements
Qi Zhou acknowledges the co-op program provided by the Science and Technology Division of Corning Inc., which allowed her to conduct this study. The authors acknowledge financial support for this research provided by the National Science Foundation under Grants No. DMR- 1944510 and DMR-1928538.
About the authors:
Qi Zhou completed her Ph.D. in winter 2022 under the advisement of Mathieu Bauchy at the University of California, Los Angeles, and Ying Shi at Corning Inc. Contact Zhou at qi1197@ucla.edu.
Editor’s note: Zhou will present the 2023 Kreidl Award Lecture at the Glass & Optical Materials Division Annual Meeting on June 6, 2023. Learn more about the conference.
One potential way to reduce carbon emissions from soda ash is to use the Solvay process with CO2 derived from a CCUS stream rather than calcined limestone.
